Korrektur der Selbstwechselwirkung in der Theorie der elektronischen Struktur fester Stoffe
Forschende des NOMAD-Labors am Fritz-Haber-Institut der Max-Planck-Gesellschaft, zusammen mit Kolleg*innen der Fudan-Universität in Shanghai, haben eine Methode entwickelt, um einen bekannten Fehler in der Dichtefunktionaltheorie (DFT) zu korrigieren. Dieser Fehler führt oft zu ungenauen Vorhersagen über die elektronischen Eigenschaften von Materialien, was die Entwicklung neuer Materialien erschwert.

Der sogenannte Selbstwechselwirkungsfehler beeinflusst oft die Genauigkeit von DFT-Berechnungen. Um dieses Problem anzugehen, entwickelte das Team eine neue Methode, die auf dem Edmiston-Ruedenberg-Formalismus basiert, der diesen Fehler korrigiert und nicht von den numerischen Instabilitäten betroffen ist, die seit ihrem ursprünglichen Vorschlag durch Perdew und Zunger im Jahr 1980 bisher vorgeschlagene Korrekturschemata beeinträchtigt haben.
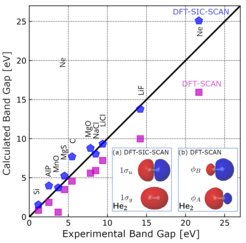
Der vorgeschlagene Ansatz verbessert die DFT-Vorhersagen für Schlüsseleigenschaften wie Ionisationspotentiale und Bandlücken in Molekülen und Festkörpern erheblich. Zum Beispiel wird der durchschnittliche Fehler bei Bandlücken-Vorhersagen von 31,5 % auf 18,5 % reduziert. Dies ist ein wichtiger Schritt, um die Zuverlässigkeit von Computersimulationen für die Materialforschung zu erhöhen.
Dr. Sheng Bi betont, dass diese Methode zeigt, wie wichtig die Korrektur des Selbstwechselwirkungsfehlers für wesentlich genauere Berechnungen ist. Obwohl der Ansatz nicht in allen Bereichen überlegen ist, ebnet er den Weg für die Entwicklung neuer, verbesserter und somit präziserer Korrekturschemata für die Materialwissenschaft.
Die Ergebnisse wurde im Journal of Chemical Physics im Artikel „Self-interaction corrected SCAN functional for molecules and solids in the numeric atom-center orbital framework” veröffentlicht.











