Mit aktivem Lernen zu neuen Solarzellen

Wie kann man sich auf etwas vorbereiten, ohne zu wissen, was es sein wird? Wissenschaftler*innen des Berliner Fritz-Haber-Institutes und der TU München haben sich dieser geradezu philosophischen Frage im Kontext des maschinellen Lernens gewidmet. Lernen ist eigentlich nichts anderes als das Zurückgreifen auf gemachte Erfahrungen. Um mit einer neuen Situation umgehen zu können, muss man vorher halbwegs ähnliche Situationen erlebt haben. Beim maschinellen Lernen bedeutet dies, dass man dem Lernalgorithmus entsprechend viele Daten zur Verfügung stellt. Was aber, wenn es so unendlich viele Möglichkeiten gibt, dass es schlicht unmöglich ist, für alles ähnliche Daten zu generieren?
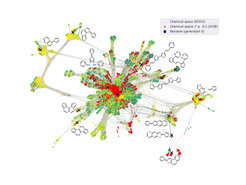
Genau dieses Problem ergibt sich sehr oft bei der schier endlosen Vielzahl von möglichen Molekülen. Organische Halbleiter bilden die Grundlage für so zukunftsträchtige Anwendungen wie tragbare Solarzellen oder zusammenrollbare Bildschirme. Hierfür müssen aber noch bessere organische Moleküle gefunden werden, aus denen sich diese Materialien zusammensetzen. Für solche Suchaufgaben werden zunehmend Verfahren des maschinellen Lernens eingesetzt, die entweder mit gerechneten oder gemessenen Daten trainiert werden. Allerdings wird die Anzahl grundsätzlich möglicher organischer Moleküle auf ungefähr 1033 geschätzt – eine unfassbar große Zahl, die es unmöglich macht, einfach so Daten zu erzeugen, die diese riesige Vielfalt halbwegs abdecken. Zumal die allermeisten Möglichkeiten komplett unbrauchbar für organische Halbleiter sind und es sprichwörtlich gilt, die Nadel im Heuhaufen zu finden.
In ihrer in Nature Communications erschienenen Arbeit gehen das Team um Prof. Karsten Reuter, Direktor der Abteilung Theorie am Fritz-Haber-Institut, dieses Problem mit sogenanntem aktiven Lernen an. Anstatt mit vorhandenen Daten zu lernen, bestimmt dieser Lernalgorithmus sukzessive selbst, welche Daten er braucht. So berechnen die Wissenschaftler*innen mit aufwändigen Computersimulationen erst einmal für eine Anzahl kleinerer Moleküle elektrische Leitfähigkeitsdaten, die eine Eignung in organischen Halbleitern und Solarzellen andeuten. Basierend auf diesen Daten überlegt sich der Algorithmus, ob kleinere Modifikationen der Moleküle entweder zu sehr guten Eigenschaften führen oder ob er sich unsicher über diese Eigenschaften ist, weil ihm ähnliche Daten fehlen. In beiden Fällen fordert er automatisch neue Simulationen an, verbessert sich anhand der so generierten Daten, überlegt sich neue Moleküle – und so geht dies kontinuierlich weiter. In ihrer Arbeit zeigen die Wissenschaftler*innen, wie effizient auf diese Weise neue vielversprechende Moleküle gefunden werden, während sich der Algorithmus immer weiter durch die Weiten des molekularen Raums fräst, sogar genau jetzt in diesem Moment. Jede Woche schlägt er neue Moleküle vor, die die nächste Generation von Solarzellen einläuten könnten, und er wird immer besser.











