
Photo-Electrocatalysis

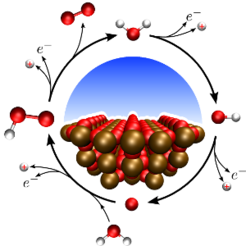
Especially in an energy context, electro- and photo-electrocatalysis assume a more and more central. Large scale sustainable generation of energy carriers -- such as H2 and Methanol -- and at the same time reduction of CO2 are important targets in light of climate change and finite fossil resources. The feasibility of such an approach obviously rests mainly on the catalysts, their stability, availability and efficiency. In order to optimise existing catalysts and find completely new materials, an atomistic understanding of the specific reaction mechanics is of paramount importance and it is there that computational approaches can contribute to catalysis research. I our group, we study the pathways of common electrocatalytic reactions such as e.g. Water oxidation for H2 production and specifically test common assumptions and simplifications. To allow for an unbiased examination of pathways, that for example also includes defected catalyst surfaces and charged reaction intermediates, we develop embedding methods which remove finite size effects inherent to standard periodic-boundary condition simulations. Finally, we also investigate the influence of the solvent on reaction properties through a mixture of explicit and implicit solvation methods.