Correcting Self-Interaction in Solid-State Electronic Structure Theory
Researchers from the NOMAD Laboratory at the Fritz Haber Institute of the Max Planck Society, along with colleagues from Fudan University in Shanghai, have developed a method to correct for a well-known error in density-functional theory (DFT). This error often leads to inaccurate predictions about the electronic properties of materials, complicating the development of new materials.

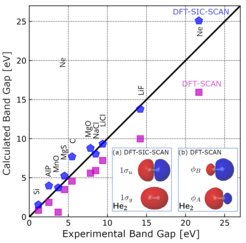
The so-called self-interaction error often affects the accuracy of DFT calculations. To address this issue, the team developed a new method based on the Edmiston-Ruedenberg formalism that corrects for this error and that is not affected by the numerical instabilities that have affected previously suggested correction schemes since their original proposal by Perdew and Zunger in 1980.
The proposed approach significantly improves DFT predictions for key properties such as ionization potentials and band gaps in molecules and solids. For instance, the average error in band gap predictions is reduced from 31.5% to 18.5%. This is an important step in increasing the reliability of computer simulations for materials research.
Dr. Sheng Bi emphasizes that this method demonstrates how correcting the self-interaction error is key to much more accurate calculations. Although the approach is not superior in all areas, it paves the way for the development of new, improved, and hence more precise correction schemes for materials science.
The results were published in the Journal of Chemical Physics in the article ,,Self-interaction corrected SCAN functional for molecules and solids in the numeric atom-center orbital framework".











