
Thermodynamic description of electrified interfaces
Electrochemical interfaces
Electrochemistry deals with the processes at the interface between an electron conductor and an ion conductor. By application of a potential, electronic surface charges aggregate at this interface inducing possibly electrochemical reactions, e.g. at a metal-water interface. As these involve electron transfer processes, it is typically electronic structure methods that are needed to simulate these atomistic processes. The sluggish kinetics e.g of an ion conducting liquid such as water and the shere size of the diffuse double layer however often hamper a straight forward fully explicit description.

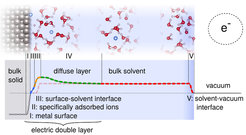
Fig. 1 sketches the different relevant parts for e.g. a metallic electrode in water: The metallic surface (I) is possibly covered with charged ions and/or molecules(II). Solvent molecules close to the surface can have an impact on the interfacial potential drops and the energetics via surface-specific orderings and charge-transfer(III). At equilibrium, counter ions accumulate in the diffuse layer (IV) with a total charge that balances all surface “excess” charge within the inner layers. The structure of this layer strongly depends on electrolyte concentration as does the induced potential difference with respect to the bulk of the solution. In the bulk of the solution, the potential is constant as the mobile ions screen any field and there is a final electrostatic potential jump at the solution-vacuum interface (V).
In the ideal case when the solvent response in regions (III-IV) is transfereable between systems, it makes sense to not include these (generic) degrees of freedom explicitly in atomistic simulations, but to use e.g. an implicit solvent, mean-field-like, continuum model to approximate their impact on the interfacial energetics.
Implicit solvent models
In recent years, the coupling of such implicit solvent models to density functional theory codes, has indeed enabled to capture at least phenomenologically the solvent and electrolyte response. It turns out that such an approach can rationalize much richer behaviours for interfacial processes than more traditional simulations that neglect the impact of capacitive charging: Among these are e.g. Non-Nernstian effects of the pH, as often observed in experiments (Refs. [1,2]).
Most of these implicit models, depend on a limited range of parameters, the most important one being the chosen onset of the dieelectric. Our go-to model is the SCCS model by Andreussi and coworkers, where the dieelectric onset is determined self-consistently based on the electronic charge density, for which accurate interfacial capacitances (and partly potentials of zero charge) are obtained when using optimized parameters (Ref.[1]).
However, to date, no general agreement exists, how the accuracy of such models can be compared, and even evaluated.
Recently, we worked on understanding better the effect of fields on the prediction of cyclic voltammograms, which we hope in the future will help to perform such much-needed assessments e.g. via comparison to experiments (Ref. [2]).
Furthermore along the lines of these studies we were able to gain a better understanding of the relation of the traditional "Computational Hydrogen Electrode" approach and a fully grand canonical description of electrochemical interfaces. As a results we were able also to recover classical electrochemical descriptors, such as the electrosorption valency, which we can now assess computationally (Ref.[2,3]).
Research outline
As implicit models enable the use of varying the applied potential, as compared to systems without a proper description of the solvent, we follow two pathways in our group:
First, we are still in the process of assessing the effect of such implicit models on the interfacial energetics, and trying to develop protocolls to evaluate the accuracy of such predictions, e.g. via comparison to experimental observables.
At the same time, in many systems the interfacial water properties are so specific, that an all-implicit solvent description fails. For such systems and in order to improve our general understanding, we have started to study electrochemical interfaces using a fully explicit description.
Updates will follow soon.
References
[1] Grand canonical simulations of electrochemical interfaces in implicit solvation models
NG Hörmann, O Andreussi, N Marzari, J. Chem. Phys. 150, 041730 (2019)
[2] Electrosorption at metal surfaces from first principles
NG Hörmann, N Marzari, K Reuter, npj Comput. Mater. 6, 136 (2020)
[3] Thermodynamic Cyclic Voltammograms Based on Ab Initio Calculations: Ag(111) in Halide-Containing Solutions
NG Hörmann, K Reuter, JCTC (2021)