The chemistry of electrocatalytic oxygen evolution over Iridium
Juan-Jesús Velasco-Vélez, Hong Nhan Nong#, Lorenz J. Falling, Arno Bergmann$, Malte Klingenhof#, Hoang Phi Tran#, Camillo Spöri#, Rik Mom, Janis Timoshenko$, Guido Zichittella§, Axel Knop-Gericke, Simone Piccinin&, Javier Pérez-Ramírez§, Beatriz Roldan Cuenya$, Peter Strasser#, Detre Teschner, Travis E. Jones

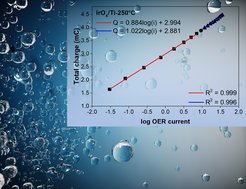
The oxygen evolution reaction (OER) plays a decisive role in many alternative energy schemes as it supplies the protons and also the electrons required for converting renewable electricity into chemical fuels. Electrocatalysts accelerate the reaction by facilitating both the required electron transfer and the formation as well as rupture of chemical bonds. This involvement in fundamentally different processes results in complex electrochemical kinetics that can be challenging to understand and control, though reaction kinetics typically depends exponentially on overpotential. Such behavior emerges when the applied bias drives the reaction in line with the phenomenological Butler-Volmer theory that focuses on electron transfer, making it possible to use Tafel analysis to gain mechanistic insight under quasi-equilibrium or steady-state assumptions. But the charging of catalyst surfaces under bias also affects bond formation and rupture, and the impact of this on the electrocatalytic rate is not accounted for by the phenomenological Tafel analysis and often unknown. Here we report pulse voltammetry, operando X-ray absorption and photoelectron spectroscopy (XAS, XPS) measurements together with DFT calculations on iridium oxide to show that the applied bias does not act directly on the reaction coordinate, but affects the electrocatalytically generated current through charge accumulation in the catalyst. [1,2] We find that the logarithm of the rate of OER linearly correlates with the charge accumulated. The applied potential drives the formation of empty Ir 5d states, ascribed to formally Ir5+ species, and the concomitant appearance of electron-deficient oxygen surface species (μ1-O and μ2-O) that are responsible for water activation and oxidation. Potentiodynamic XAS probing the electron-deficient oxygen species reveals a linear correlation between charge and μ2-O coverage suggesting that oxidative charge is accumulated by surface deprotonation (coupled to electron transfer). DFT calculations indicate that water nucleophilic attack on a surface oxyl (μ1-O) is rate-limiting. O–O coupling occurs with concerted transfer of H to a μ2-O site. The activation free energy (Ea) for O–O coupling obeys the Brønsted–Evans–Polanyi relationship, which is familiar from traditional catalysis, with Ea depending linearly on ΔHrxn, which in turn is controlled by oxidative (rather than capacitive) charge. Ea of this elementary step decreases linearly with the amount of oxidative charge stored, giving rise to the above-mentioned rate dependence on accumulated charge. According to classical electrochemical theory, such behaviour is consistent with inner-sphere reactions, in which the reacting species adsorbed on the electrode surface are insensitive to any double layer, and inconsistent with outer-sphere reactions involving weakly interacting species far from the electrode surface that obey Butler–Volmer kinetics. Demonstrating how inner-sphere chemistry controls electrocatalytic OER rates establishes a fundamental link between thermal catalysis and electrocatalysis that enables tools and concepts of traditional catalysis to be applied to electrocatalysis.
References
[1] H.N. Nong, L.J. Falling, A. Bergmann, M. Klingenhof, H.P. Tran, C. Spöri, R. Mom, J. Timoshenko, G. Zichittella, A. Knop-Gericke, S. Piccinin, J. Pérez-Ramírez, B.R. Cuenya, R. Schlögl, P. Strasser, D. Teschner, T.E. Jones, Key role of chemistry versus bias in electrocatalytic oxygen evolution, Nature. 587 (2020) 408–413. https://doi.org/10.1038/s41586-020-2908-2.
[2] J.-J. Velasco-Vélez, E.A. Carbonio, C.-H. Chuang, C.-J. Hsu, J.-F. Lee, R. Arrigo, M. Hävecker, R. Wang, M. Plodinec, F.R. Wang, A. Centeno, A. Zurutuza, L.J. Falling, R.V. Mom, S. Hofmann, R. Schlögl, A. Knop-Gericke, T.E. Jones, Surface Electron-Hole Rich Species Active in the Electrocatalytic Water Oxidation, J. Am. Chem. Soc. 143 (2021) 12524–12534. https://doi.org/10.1021/jacs.1c01655.
# Department of Chemistry, Chemical and Materials Engineering Division, Technische Universität Berlin, Germany
$ Department of Interface Science, Fritz-Haber-Institute of the Max-Planck-Society, Berlin, Germany
§ Institute for Chemical and Bioengineering, ETH Zurich, Zurich, Switzerland
& Istituto Officina dei Materiali, Consiglio Nazionale delle Ricerche, CNR-IOM, Trieste, Italy