Hydrated Electrons at Electrochemical Interface
. Reprint with permission from ACS publications.")
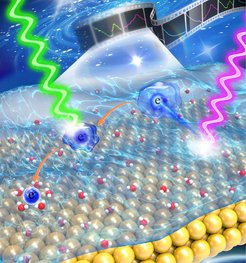
Electrochemical reactions are omnipresent in most people’s everyday lives, even if they don’t know it - charging a mobile phone, for example. When doing that, electrons are sent to a solid/electrolyte interface to store chemical energy at the electrode which can be used later on when the device is used. To improve such electrochemical devices, the fundamental processes that are involved at such interfaces need to be understood. The challenging questions is: How does the electron interact with solvent molecules as it transfers from the solid electrode into the liquid phase solution? This question turns out to be non-trivial even though metal/aqueous solution interfaces are ubiquitous in many electrochemical systems.
The challenges are twofold. For one, the electron transfer takes place at a solid/liquid interface, where both adjacent phases are condensed matter, which prevents the use of many of well-developed surface science techniques (as those are often based on the detection of electrons or ions in vacuo). This highlights the need for a technique to probe the structure and dynamics of the electron. Secondly, these processes are believed to be extremely fast (on the order of tens of femtoseconds to picoseconds, a time range in which at light speed one could not even travel a millimeter). While conventional electrochemical tools such as potentiostats (devices developed to drive electrochemical reactions) are good at counting the number of electrons during a reaction, they are not fast enough to follow ultrafast dynamics such as electron solvation.
To track the ultrafast dynamics of electron hydration at the metal/aqueous solution interface, a research group led by Dr. Yujin Tong at the Physical Chemistry Department developed a novel optoelectronic sampling scheme, in which ultrafast laser pulses were combined with an electrochemical recording system. This allows the ultrafast electron hydration dynamics (i.e. the interaction of electrons with water molecules) to be followed in real time.
The concept is similar to that shown in the cover article: a combination of two femtosecond laser beams induces a photovoltage that can be simply read out by a commercial potentiostat. Controlling the time delay between the two ultrashort laser pulses, a “movie” about electron solvation can be shot.
“The properties of the solvated electron at the gold/aqueous solution interface are significantly different from those in the solution bulk. The interaction with the metal electrode influences both the energy and lifetime of the different transient states of the hydrated electron,” describes Dr. François Lapointe, a former postdoc at the FHI and now a permanent Research Officer at the National Research Council Canada. “More intriguing are the structural properties of these different states of the hydrated electron, the species change from completely isotropic (similar to an ideal sphere) to fully anisotropic (like a stick) during their relaxation process. Such information will be important for understanding photo-electrochemistry at the metal/solution interface,” he adds.
This study also suggests it should be possible to trigger electrochemistry on femtosecond scales using the same methodology: ultrafast charge transfer can be initiated using a pump pulse of appropriate photon energy (e.g., UV) and the second push pulse can be used to induce a photovoltage or photocurrent to monitor the reaction. As such, the novel detection scheme developed in this study opens a wide parameter space (the nature of solvent and electrolyte, temperature, local potential, etc.) to investigate the molecular level structural changes during charge transfer. Furthermore, it is expected that such optoelectronic measurement scheme will become useful in the study of hot electron photocatalysis and plasmon-assisted catalysis, hence help the design of better solar cells.











